Full HTML
Huntington’s disease and Wilson’s disease in Sweden have a common origin in the 16th century and expanded due to effective family size of the founder family - Volume 3 Issue 1 (Jan-June), - (6 Months )
Pages: 1-17
Category: Original Research
Published Date: 22-05-2026
K Sigvard Olsson1Olof Stenum2Olof Wålinder3, Samrat RoyChowdhury4, Ruma Raha-Chowdhury5,6
Author Affiliation:
1 Department of Medicine and Clinical Nutrition, University of Gothenburg, Sweden;
2 Independent Researcher;
3 Department of Medicine, Sweden;
4 Department of Medicine, Kettering General Hospital, Kettering, UK;
5 Department of Clinical Neurosciences, University of Cambridge, Cambridge, UK;
6 Department of Psychiatry, University of Cambridge, Cambridge, UK
Correspondence: Ruma Raha-Chowdhury, Department of Psychiatry, University of Cambridge, Cambridge, UK
Keywords:
Effective family size, Huntington´s disease, Wilson´s disease, autosomal recessive deafness (ARD), Hereditary haemochromatosis, Long QT Syndrome, Jervell and Lange-Nielsen syndrome (JLNS), river valley populations, Vikings, Church records
Full Text:
River valley populations of northern Sweden are well suited for research of monogenic diseases due to the wilderness between them that creates sub-isolates [1], and availability of excellent church records [2]. These have been helpful in studies of recessive disorders, like hereditary haemochromatosis (HH) common in the region of Jämtland (Figure 1) due to local founder effects [3,4]. It is caused by the HFE/p.C282Y mutation that in homozygotic carriers have a risk for iron overload, particularly affecting the liver and other organs. A deep HFE pedigree analysis revealed that in some families, Wilson´s disease (WD) also segregates with the HFE. WD is like HH, another recessive inherited disorder, causing damage to the liver, and the brain (hepatolenticular degeneration), where instead of iron, copper accumulates in the liver and the brain as a result of defective biliary copper transport [5]. WD is a rare disorder caused by ATP7B mutations, and extensive pedigree studies have traced nine WD families to a common origin in a family born around 1620 [5], living in a village located around the southern branch of the Mo river (red symbol in Figure 2). The deep HFE pedigree [4] also included the Swedish long QT syndrome (LQTS) mutation [6], which in homozygotes causes concomitant hearing loss, termed the Jervell and Lange-Nielsen syndrome (JLNS) [7].
This syndrome was first identified in a HH family where two male siblings were born deaf [8]. However, as tests for LQTS and GJB2 mutations were negative in this family, a whole exome sequencing (WES) was performed. Results showed autosomal recessive deafness (ARD) caused by homozygous mutations of the WHRN/DFNB31 gene. Through extensive genealogic studies, the deafness could be traced to a common origin in a family born around 1579–1580 of village K8. From this family with effective family size (EFS) assessment, it was found that there was a marital spread of deafness upstream of the Angerman river (Figure 2) by early colonizers of the Lapland habitat in the region of Västerbotten (Figure 1). This could be associated with selective advantages of HH gene [4], and in agreement with upstream observations from river Saguenay (SLSJ) of Quebec [9].
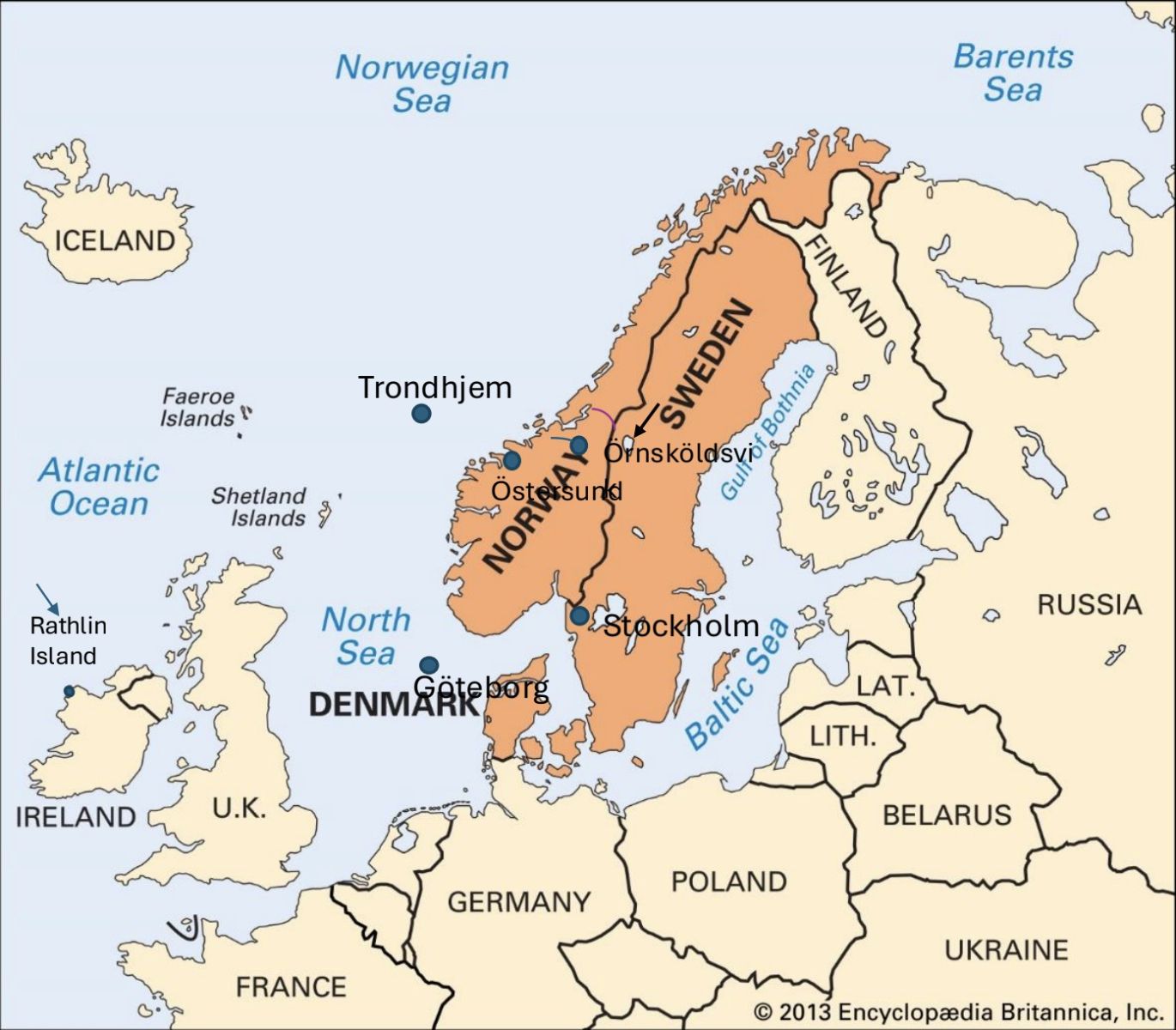 |
Figure 1. Map of the countries surrounding the Norwegian Sea, including the study area in central Sweden. The study region is indicated by an arrow located northwest of Örnsköldsvik, along the Gulf of Bothnia.
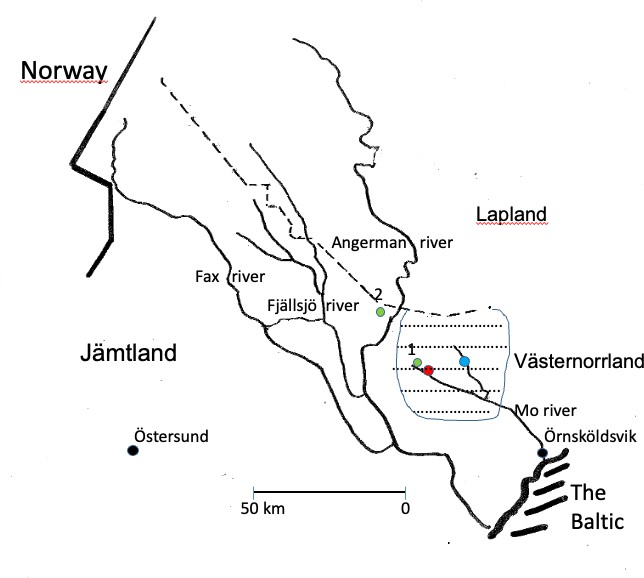
Figure 2. Enlarged view of Figure 1, illustrating Parish A (63.27?N) situated upstream along the Mo River in the Västernorrland region of Sweden. The residences of Huntington disease families investigated during 1933–1934 are indicated by green symbols. The origin of the Wilson disease founder family (circa 1620) is marked in red, whereas the founder family associated with autosomal recessive deafness (circa 1579–1580) is indicated in blue.
2. Aims
The present study firstly was to test whether HD and WD had a common origin as reported in church records. Secondly, whether their evolution was influenced by the EFS in 1579-1580 founder family.
3. Methods
Demographic studies of church records (digitally available today) in which affected patients were searched for in a catechetical registry of “sinnessjuka och idioter” 1908-1923 (insanity and fools) Swedish National Archives Registry IIa2a were carried out. This registry contained the names of 62 individuals, 51 of them from the Mo River population (Figure 1). We also studied parish records of the death and funerals Swedish National Archives Registry. Valuable information about the local population history was obtained from “Roots in Anundsjö” [16].
The previous study had been approved by the Regional Ethical Review Board at the University of Göteborg, (Dnr 834/14, T 214/16 and T1076-16). Subjects affected with HD and WD were included in our haemochromatosis HH/WD database Holger 7 and traced to founders. Also included were 51 individuals from the “A-skog” church archives (Figure 2). This resulted in an increase in the database from (n =13720 to n=16216) [8]. Pedigrees were drawn with Cyrillic 2.1 software (Cyrillic Software, Old Beaconsfield, UK), as previously described [4]. Ethical questions were evaluated and after appropriate information, the new WD patient gave written informed consent. One new HD patient is no longer alive like his younger brother and both parents are dead, none of them could sign a consent.
5.1. Three hypotheses were examined
- Is the origin of HD and WD common with other recessive disorders due to interbreed within a small population?
- Is the origin of HD and WD related to the 1579–1580 founder family and spread through EFS?
5.2. Most recent common ancestor of HD and WD is a family born 1660-1661
The author, Dr Torsten Sjögren, in 1936, first visited a small village in “J-skog” (indicated with green symbol, west of Angerman river,(Figure 2), and the results are shown in the pedigree of Figure 3 (left part). He had investigated a number of patients (lower left arrows, in (Figure 3), all with the descent from III:2 (upper arrow), a son of a farmer II:3, born 1749 with HD according to unanimous sources. His daughter III:3, lived in parish “A-skog” (green symbol, Figure 2) and her family was studied one year later (in April 1937), and the results are shown in (Figure 3). This pedigree seems to include a common founder effect from a marriage of III:1 OP, born 1660 and III:2 MJ, born 1661 (red symbols in the centre of the pedigree in (Figure 4). She was a granddaughter of I:1 AP and - I:2 KN (1620?) known as the WD/HH founder family5, which included ten WD families (red symbols in the centre of the pedigree, Figure 4) and 36 (90 Percent) of the 40 HD families (green symbols) in Figure 4. However, the author was uncertain about the origin of a man born 1757 of Family 27 (Figure 4), and another man born in 1799 of Family 36 (Family 37,38,39, Figure 5) and the origin of the father born 1794 (arrow, (Figure 5) of Family 34. He was executed in 1845 for murdering his wife and seven of his children on August 12, 1843, indicating some degree of mental instability.
Two children survived, one with HD (Family 34) having three affected children (Family 35), the other a grandson born in 1892 (green star, gen X, (Figure 5), whose details was found in the 1908-1923 catechetical ID registry (ID number 28), a batsman (naval soldier, and his story is noteworthy). He was a batsman, who fell ill in Stockholm in 1913 and was admitted to a mental hospital. (The catastrophic events of his family were depicted on the front page of his case record).
He was further referred to Frösö mental hospital (close to Östersund) in 1915, dis-charged (not free from symptoms) in 1918, and died (committing suicide) in 1927. To the left of the batsman is a woman of Family 30, who emigrated with her children to the USA. She was in the descent of woman born in 1798, of Family 28, and was severely affected by HD and gave birth to 18 children of whom seven were reproducing, an example of EFS (Families 29,30,31,32,33). Another new HD patient born in 1952 (green symbol, Figure 5) and his brother (who committed suicide) had inherited the effected gene from their mother born in 1924, who was the descent of Family 29. The total number of suicides was 19, only 10 are shown (lower arrow, Figure 5). Eighteen family members died in mental hospitals (most often from tuberculosis). The author reported one HD patient with deafness (may be carrying WHRN/DFNB31 gene), she is shown by a lower arrow (Figure 5). Church records reported two additional HD cases marked by green stars such as X:9 and XI:1 (Figure 5) with “danssjuka” the Swedish word for HD. The pedigree also included 29/51 (57 Percent) individuals in the 1908-1923 catechetical ID registry, only some of those cases are shown (black symbols, Figure 5).
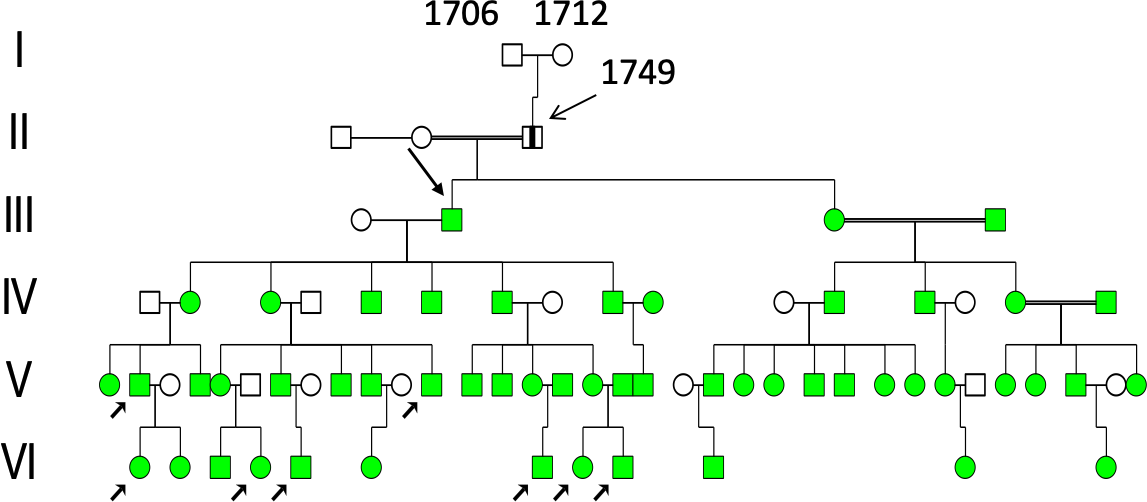 |
Figure 3. Pedigrees of Huntington´s disease (HD) patients investigated in 1933. Individual II:3, born (b) in 1749, was reported to be affected by HD. His son, III:2 (b 1778), left Parish A (green symbol 1 in Figure 2) to become a pioneer settler in a nearby parish (green symbol 2 in Figure 2), thereby creating a local founder effect. The right section of the pedigree was investigated in 1934 when the author returned for additional studies. HD patients are indicated by filled green symbols, whereas individuals personally investigated by the author are marked with lower-left arrows.
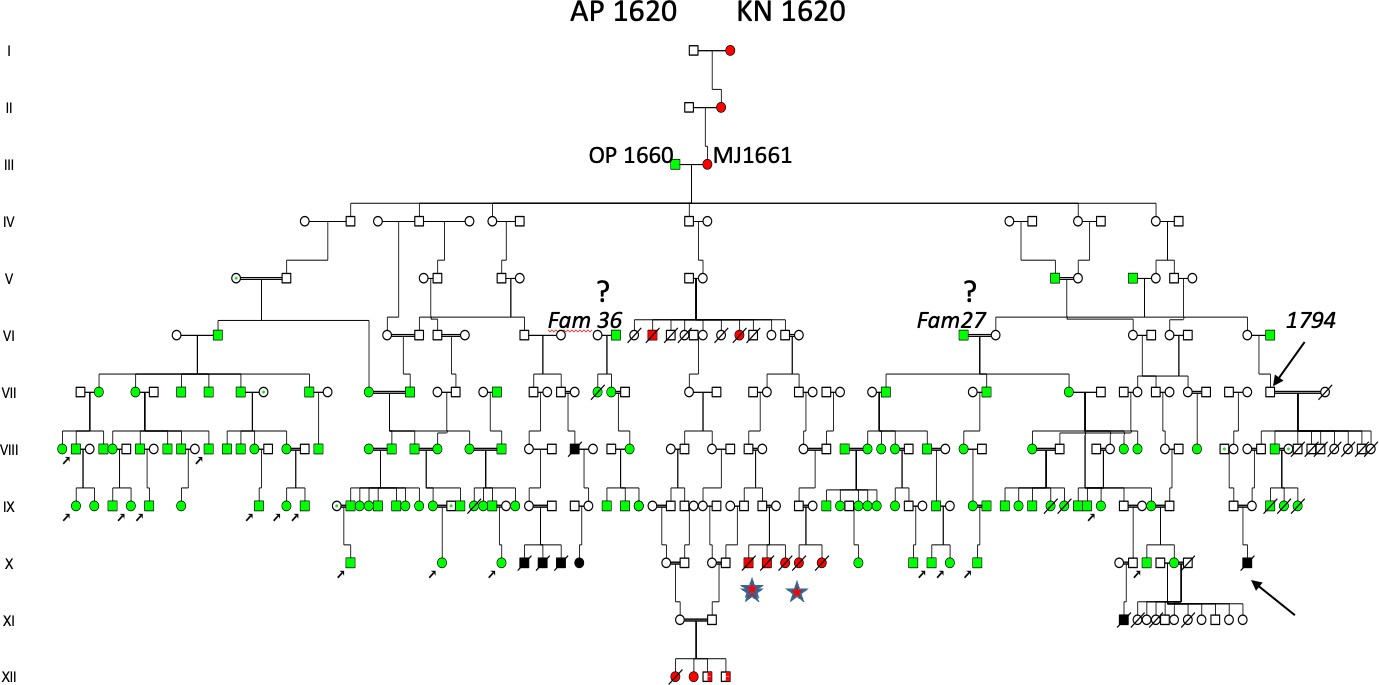
Figure 4. Pedigree of HD patients investigated during 1933–1934 and traced to a common origin in individuals III:1–2, where III:2 (born in 1661; marked in red) was a granddaughter of AP 1620–KN 1620, recognized as the Wilson disease (WND) founder family [8]. This lineage includes 36 (90%) of the HD families, excluding Family 36 and Family 27. The author also expressed uncertainty regarding VII:25 (b 1794; upper arrow), who killed his wife and children. Two descendants survived, and X:28 (a båtsman, i.e., naval soldier; lower-right arrow) developed illness at 21 years of age. His 1913 hospital record documents the catastrophic events affecting his maternal family. Individual X:17, who married twice, emigrated with children to the United States. Individuals identified through church records are represented by black symbols. At the center of the pedigree is a descendant family with Wilson´s disease (red symbols), of which two families (indicated by stars) were identified in church records.
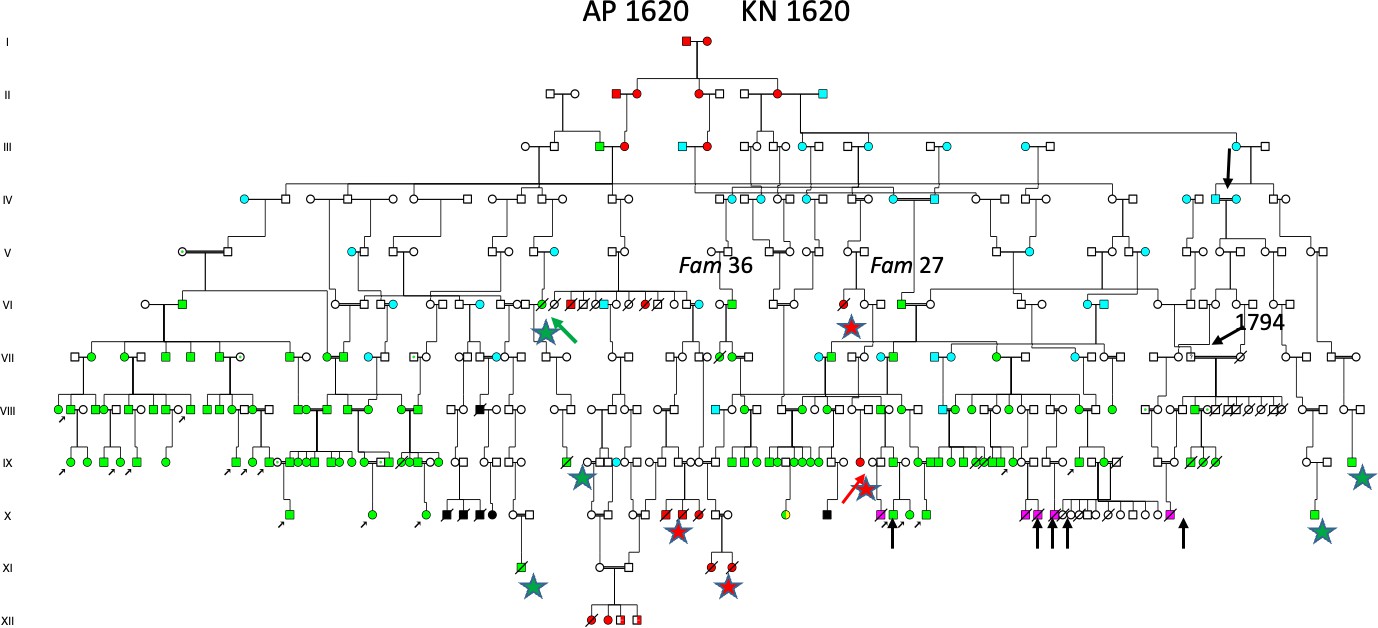
Figure 5. Extended pedigree from Figure 4, including Families 27 and 36, all descending from the AP 1620–KN 1620 founder family [8]. The pedigree includes five HD cases identified in church death and funeral records (green stars), among whom VI:14 (green arrow) was reported to have frozen to death at 46 years of age. Two additional WND cases are marked with red stars, including one individual (red arrow) who died at 17 years of age at Frösö mental hospital. Suicidal outcomes, including that of the båtsman, are indicated by lower arrows. Blue symbols denote descendants of the 1579 S & 1580 K family residing at the blue-marked location in Figure 2. One individual (upper arrow) in generation IV was recognized from the hearing-loss family [8], characterized by a high effective family size (EFS), with 8 of 14 children reproducing.??????
5.3. Additional Wilson disease (WD) gene in the extended HD/WD pedigree
From pedigree studies (Figure 5), it was indicated that ATP7B mutations of WD gene was carried by MJ (1661) III:2 as previously described. A new WD patient[5] was seen as XI:V (Figure 5), who inherited the p.H1069Q mutation from her mother. There were additional four Wilson families found in church records (marked with red stars), another example being IX:33, a five-year-old boy who died with vattusot (ascites), like his uncle VIII:28 at age 46. A 57-year-old farmer, X:20, suffered from severe anaemia (Hb 22%), icterus, and ascites that required paracentesis to remove excess fluid (2500 cc) as treatment. He died in 1933 after four days in hospital from “Hepatitis acuta” [17]. His paternal aunt born in 1836 (red star, generation IX, Figure 5) died from “vattusot” or ascites in 1866. His paternal aunt born in 1836 (IX:24) died from “vattusot” at age 30. In the next generation there were WD families previously described[4,5], also including the family XI:29, XI:30, and XI:31 (red star, Figure 5). XI:29 was an eight-year-old boy with icterus and ascites who died after four days in hospital from “Hepatitis syphilitica”. His 18-year-old brother XI:30 was hospitalized for “psychosis and albuminuria” and died three years later. Their sister (XI:31) died at age 15 in 1931 from “nephritis and ascites”. A first cousin marriage (XI:27–XI:28) resulted in the pedigree previously described[4,5], in which three siblings died from WD. To their right were two sisters, XII:23 and XII:24 (red star), who both died from “Cirrhosis hepatis c ascites”; the first at age 10 in 1938 and her sister in 1945 at age 14. Another example of WD in this family was the diagnosis of “Hepatitis syphilitica” in an 8-year-old XI:20 and XI:21 with “psychosis and albuminuria” and slurred speech, having neurologic Wilson’s disease followed by death at the age of 12. These findings indicated that a spectrum of hepatic disorders was affected due to WD gene inheritance, that co-inherited in HD families.???
5.4. HD and WD can be traced to the 1579-1580 founder family and expanded due to effective family size (EFS)
The two missing families of (Figure 4) Family 27, and Family 36 (37,38,39) had a common origin in a woman born in 1695, marked by a red arrow in (Figure 5). She was a daughter of II:8 born in 1649, the youngest daughter of the AP-KN born in 1620 (Figure 5) in the founder family and married to II:9 born in 1646, (a son of I:3 SM born in 1620). In the descent of the II:8–II:9 family were not only Fam 27 and Fam 36, but additional HD Families 20–40, which indicated that 15 were in coinheritance with members of the first founder family of the Figure 4 pedigree. All 40 HD families had origin in the I:1–I:2 WD/HH founder family also including the 15 children of the family previously described[8] (Figure 5), now marked by an upper black arrow in the right part of the pedigree (generation IV, Figure 5). A new Wilson patient was included (lower red arrow), who inherited ATP7B/H1069Q mutation from her mother. XI:17 (Red star) was a young woman born in 1908 who was admitted to the Frösö mental hospital in 1923 and died on 14th of August 1925 from “Insanio e laesione cerebri”, a diagnosis replaced by “Mb Wilsoni”. In her descent was XI:1 (green star) (equal to X:3 of Figure 4) who died from HD in 1937. The man with “danssjuka” (the Swedish word for HD) XI:9 of (Figure 5) is now seen as XI:24 (green star). Not shown in the descent of SM born in 1620 (this record comprising of 88 pages of A4 size papers), large numbers died of suicides (n = 27) including the young brother of the new HD patient (green arrow). The author had been uncertain about the mother of his first HD patient, she was KN born in 1712, I:2 of (Figure 4). She was shown as IV:3 of (Figure 5) and had an origin in the pedigree of (Figure 6).
5.5. Introduction of hereditary haemochromatosis, the long QT syndrome and Autosomal Recessive Deafness (ARD) in the ancestral pedigrees
The founder family members MÖ born in 1579 and SM in 1580 are a real treasure for the genetic studies, where many generations interbred within the close relatives and introduced many other Autosomal Recessive (AR) diseases including hereditary haemochromatosis, the long QT syndrome (LQTS) and autosomal recessive deafness as previously reported. The pedigree of KN born in 1712 was shown as V:3 in the descent of II:2, a daughter of the I:1–I:2 MÖ 1579–SM 1580 founder family (Figure 6). SM was born in 1620, (Figure 5) as shown II:7 (family details see Figure 5). It was difficult to prove that maternal grandparents of the new HD patient (green arrow) were homozygotes for any other AR diseases.
The descent of SM born in 1620 (pedigree information comprises of 87 pages of A4 size papers) also includes 46 deafness families who inherited homozygous mutations of the WHRN/DFNB31 gene and haemochromatosis (HH) gene[8] (Figure 5). The Figure 6 pedigree is incomplete because it also includes hereditary haemochromatosis, the long QT syndrome and Autosomal Recessive Deafness and reported in previous publications (Figure 5).
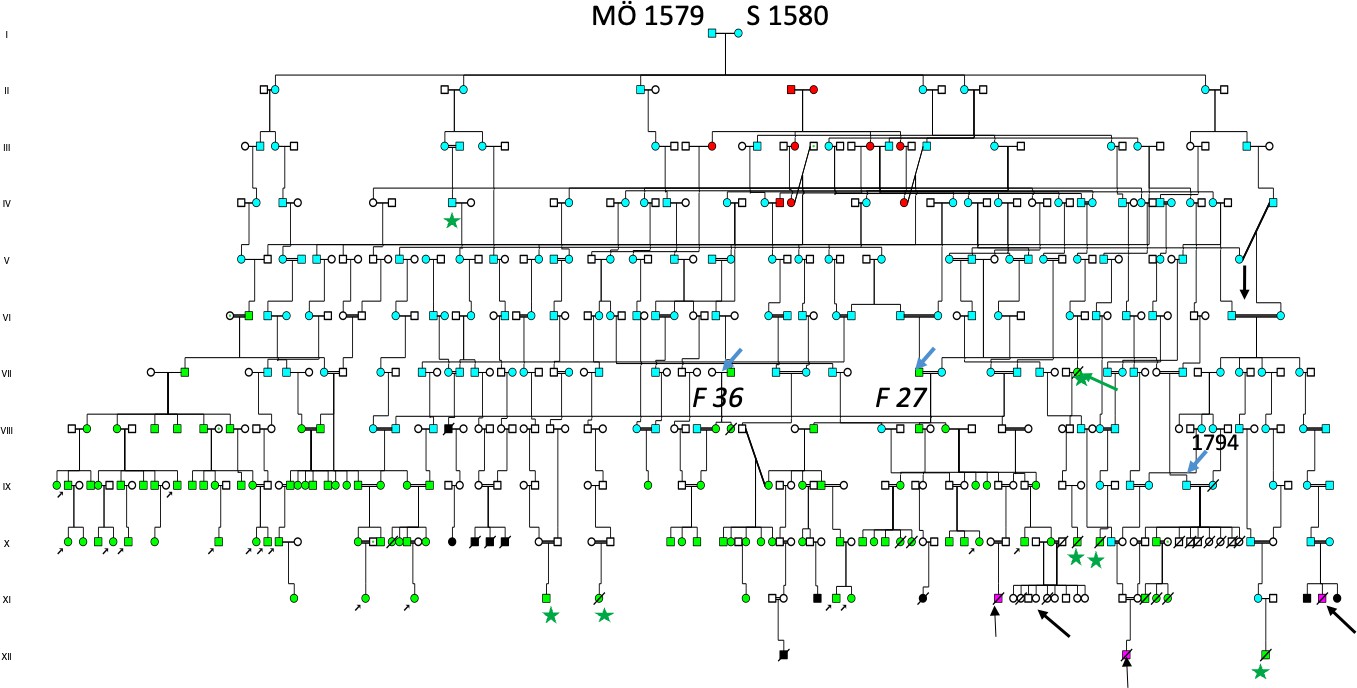
Figure 6. Pedigree illustrating the common origin of 40 HD families and an additional six families identified through church records (green stars), all traced to the founder family MÖ 1579–S 1580 living upstream along the Mo River (blue symbol in Figure 2). For simplicity, the blue color is used throughout their descendants, including the EFS family shown in Figure 5 (upper arrow). Individual IV:7 (b 1688) was described as having “fallandesot” (epilepsy) and other fragilities, originating from individuals III:5–6, a consanguineous marriage between first cousins once removed. The woman reported to have frozen to death is shown in generation VII (green arrow). Two families emigrated: one to the United States (lower-right arrow; see Figure 5) and the second family, shown to the right, emigrated to Canada.
On December 23, 1715, the church inspector reached village Hälla in Lapland (yellow symbol Figure 2), where he spent the night with a colonizer who was “dumbe” = deaf. “He was good and pious man”, with many children. Our studies suggest he is identical to Pål M born in 1680, who was of Finnish origin with permission to settle after the Lapland Bill 1670. He had 5 children reproducing, repeated in next generations when his grandson Matts born in 1751 became a colonizer of Laxbäcken (salmon stream). This village is seen upstream the main river at L of Figure 2.
Together with his wife they had 8 children, all reproducing suggesting high effective family size (EFS). Their first daughter born in 1778 came to include Fam D 24 at Laxbäcken, 1784, her sister had Fam D 87 in Mattsdal = (Matts valley) at the origin of the main river. These two families have been previously presented with different mutations in Figures 5, 6, and 7. The other siblings included Fam D14, D67, D85 and D86. The total number of deaf families from Pål born in 1680 was n = 45, upstream the main river, some of them are seen with yellow symbols in Figure 2.
In a previous study we had reported hearing loss in a 14 children family[8] (Figure 5) VI:1–2 also including two husbands IV:4 and IV:5. At that time we did not know they had Huntington´s disease HD. They are now seen in Figure 5 as dark blue symbols, sons of II:12 marked by an arrow. In their descent was the hearing loss family now including 15 children. This family is marked with an upper arrow in centre of the pedigree generation V (Figure 5).
Their first daughter A born in 1740 had HD and was the mother of Fam 34 and Fam 35. Their third daughter K born in 1744 came to marry a colonizer born in 1741 in the new habitat, which is seen in Figure 6, where they had 9 children all reproducing, suggesting high EFS. Repeat EFS was seen in next generation when their daughter MG at age 16 married a man born in 1760 from “A-skog” and had 13 children with him, 7 reproducing.
A sister of K born in 1744 was S born in 1749 who made a similar move and came to marry another colonizer born in 1754, with whom she had 10 children, 9 reproducing. Another daughter M born in 1755 married another colonizer and had with him 6 children also supporting EFS.
A man from “A-skog” born in 1714 moved to the north and married KJ born in 1722 in a new habitant (Figure 2). They had 10 children, 9 reproducing. Whether high EFS was exclusive for the 15 children family we do not know. However, a neighbour of the 15 children in “A-skog” (Figure 2) made a similar migration. He was M born in 1714, who had lost his father in 1715, and moved to marry KJ born in 1722 from Finnish colonizers. They had 10 children, 9 with EFS. He remarried and also got the same result, 10 children.
Another colonizer of Finnish origin was NA born in 1635. He was a II son of Anders born in 1610, previously described in Figures 6 and 7. They settled at village G upstream of the village (yellow symbol Figure 2) and had a considerable influence upon the small population of village “Ö” marked with a green symbol in “J-skog” close to Lapland in Figure 2.
This was the village visited by Sjögren. One stayed and came to include HD Fam 34 and Fam 35. By studying the origin PB I:5 of Figure 5 to his grandfather JO born about 1550, he is seen as I:1 of Figure 6 who had married I:2 NN, from village brown symbol (Figure 2). They had two other sons II:3 born in 1590 and II:10 born in 1587 married twice. I:1 was a priest of a neighbourhood parish married to I:2 with a son III:1 born in 1615. II:5–II:6 is the 1579–1580 founder family[8] (Figure 5).
In the descent of II:10 were 34/40 HD families and 14 WD families including both parents of the new case. One of them was Ke who married a Finnish colonizer born in 1741 and had 9 children, all reproducing, giving birth to a total of 14 HD families. Another daughter born in 1749 made a similar move and got 10 children with another Finnish colonizer born in 1754.
We have difficulties to give a new pedigree. However, most of them were previously reported in Figure 5. This finding suggested that all 40 HD cases have a selective advantage due to high EFS in early settlers of the river valley population [18].
Huntington´s disease and Wilson´s disease are well known from studies of Lake Maracaibo, Venezuela, where HD has been described in landmark studies [19,20]. The HD family was the largest genetic study ever reported, tracing through 10 generations and leading to a woman who lived in the early 19th century at Lake Maracaibo. DNA samples from the family members were analysed to map the Huntington´s gene location to chromosome 4 in 1983 and to identify the disease gene in 1993.
The patient´s samples were also used to map the Wilson disease gene to chromosome 13q in 1988 [21], which was of importance for identifying the copper transporting ATPase gene 13q14.3 in 1993 [22]. DNA samples from Swedish HD subjects were also analysed in early studies[11]. From Venezuela, Paradisi et al. also reported HD and WD with ATP7B mutations (H1069Q, R1319X, T977M) similar to those found in Sweden and as seen in Figure 7. The significance of these observations is limited considering the multitude of ATP7B mutations [23,24].
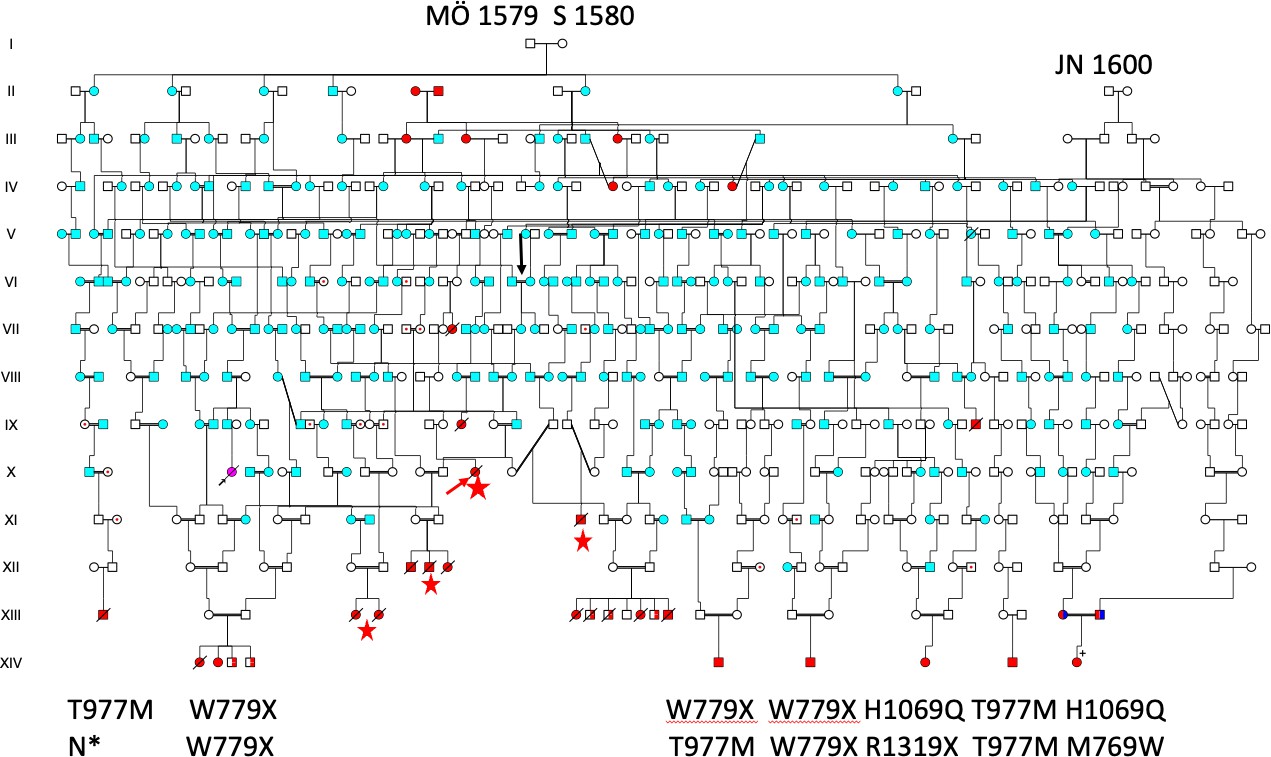
Figure 7. Pedigree demonstrating the common origin of Wilson disease (WND) patients previously described in Reference [6], including four families identified through church records (red stars). The newly identified patient XIV:9 is shown in the lower-right corner. Her parents are double heterozygotes who also carry HFE mutations (indicated in dark blue). To date, it has not been possible to connect the paternal VIII:22 ATP7B/M769W mutation to the MÖ 1579–S 1580 founder family.
There was also a clustering of HD in Jämtland (Figure 2), in agreement with HD families “living in small communities, often with suggestions that affected individuals are those who have descended from a single progenitor.” On those days, a co-segregation of two disorders was unheard of but “Typus Wilsonische Krankheit” was well known by the author, though not observed until more recently, when pedigree of Figure 4 was formed [25]. This is the first analysis that showed a common origin of the two rare neurodegenerative disorders, both originating in a founder couple III:1–III:2 born (b) 1660–1661 including 36/40 (90 Percent) of the HD probands and ten WD probands of whom three were “new” detected from church records (red stars, Figure 6).
The HD probands in generation IX or X were born between 1875 and 1893 or 225 years later from the founder couple. This would mean an average, each generation is ∼ 30 years, similar to the evolution of HD from the 1579–1580 farmer family (Figure 6) covering 10 generations through 300 years.
This evolution is slower than in Venezuela and can possibly be explained by climate differences between the altitudes N 10 and N 63.9 (see also K born 1767 of Figure 5, who was found frozen to death in November 1813) [26–28].
A weakness of the Figure 4 pedigree is the inability to show all the descent morbidity, the database covers 29 pages of A4 size paper. The author reported two HD with suicide, church records reported 16 cases and additional 15 descent members dying in mental hospitals. Author reported one HD was deaf (long arrow, Figure 4), and church records reported another 12 cases. The descent also included 29 individuals (57 Percent) from the catechetical 1908–1923 registry of “sinnessjuka och idioter” (insanity and fools). Among them was ID 28 born 1794 marked by an arrow (Figure 4) and who died from suicide. There were 10 WD families of which three were found in church records. Not shown are descent members with HH and LQTS [29].
We were unsure about which of the two III:1 or III:2 had transmitted the disease. Figure 5 gives support for an origin in the I:1–I:2 WD/HH founder family born in 1620. However, we admit uncertainty about birth dates before 1688 and I:2 might as well be born in 1610 as being suggested [16]. However, the Figure 5 pedigree showed a major influence from their neighbour K family born 1579–1580 (blue symbol Figure 2) with high EFS according to previous studies [8] (Fam 5).
Repeat EFS was first observed when SM I:3 (Figure 5) and her sons (blue marked) II:8 and III:4 were entering the pedigree. She had a total of 19 reproducing grandchildren, and her database comprises 87 pages of A4 paper to include the two missing HD families Fam 36, Fam 27 and the Fam 34 tragedy of Figure 4. We cannot exclude this tragedy from the father´s knowledge about HD in the family history (the parents were 2nd cousins twice removed from K born 1668 marked by upper right arrow of Figure 5).
A great grandchild of SM born 1620, K born 1717 (who married her 3rd cousin) was similarly unfortunate, because their five children´s descent came to include 12 suicides [30].
The Figure 6 family also included the son of II:11, E born 1632 (blue arrow generation III) also including the Fam 34 tragedy, and the three missing patients of Frösö mental hospital (Figure 5) marked by a blue arrow in generation IX (both parents originating in blue marked III:20). The Figure 6 pedigree includes a total of 26 suicides and 36 family members who died in mental hospitals (not shown). Even if suicide of young women (three were 22 years or younger) may have other causes, the high number was remarkable but in agreement with early studies [31]. The pedigree is incomplete, and we had difficulties to show all the 51 affected in the church 1908–1923 insanity registry. Only a few are seen in black symbols [32].
6.1The high numbers of disease mutations may be a reflection of EFS of the founder family
Isolation, consanguinity and inbreeding might well have been contributing factors of the present pedigrees of recessive disorders of the Mo river valley population (Figures 4, 5, and 6) like in other isolates [33,34]. However, first-cousin unions were rare, but remote consanguineous marriages were not uncommon. We believe EFS transmitted to next generations might have contributed to the expansion of the present pedigrees (Figures 5 and 6). The EFS is the number of children that reproduce in the population per reproducing woman as reported by Austerlitz and Heyer [35].
Its importance was the first demonstration of explaining the increased frequency of recessive disorders upstream of river Saguenay (SLSJ) of Quebec. The Swedish Mo river population is very small (about 4000) as compared to the SLSJ (about 300000) and the number of recessive disorders is also smaller but the effect of EFS is similar. One difference is the inclusion of dominantly inherited HD in the 16th century Swedish founder family with high EFS. A matrilineal fertility inheritance was more common in all our pedigrees (Figures 4, 5, 6, and 7), in agreement with the higher female migration rate in humans [36].
6.2. Origin of disease mutations
An import of mutations through the Baltic (Figure 1) from Finland seems likely as the hearing loss TMC1 mutation reported previously, however the hearing loss DFNB31 mutations could be more distant in origin. An import of HD and HH from Finland seems less likely as both are rare in this country. HD and LQTS [7] were first reported in Norway and at present, there are five HD centres in Norway (astri@eurohuntington.org), one of them in Tröndelag close to Jämtland where HD was first reported and recently by Roos et al [15]. (Figures 1 and 2).
The haemochromatosis (HFE p.C282Y mutation) reached a particularly high frequency in this region, possibly through an immigration from Tröndelag and their HLA haplotypes were similar. The HFE p.C282Y mutation was previously shown to reach particularly high frequencies in countries of the Norwegian Sea (Figure 1), and an older age of the mutation was proposed [45].
This was recently confirmed when a Bronze Age skeleton from Rathlin Island (arrow Figure 1) was shown to carry the HFE/p.C282Y mutation and the ancestral HLA A3B7 haplotype. The very high HFE p.C282Y frequencies (17.3 Percent) of Northern Ireland and West of Scotland (17.7 Percent [33]) were therefore not surprising [45].
The high frequency of HD in these territories was of particular interest, as shown on a map by Harper in his epidemiology of Huntington´s disease, that also included his own HD findings in Wales, further down the Norwegian Viking travel routes.
As the purpose of many Viking expeditions was no doubt to capture slaves, we cannot exclude the possibility that slaves from these territories were onboard returning Viking ships to Scandinavia [38–40]. Iceland was settled by Norsemen with Irish slaves. Whether the HTT gene migrated from Cornwall (Figure 1), Wales, Northern Ireland or Scotland is unknown.
We do not know if HFE was taken from these territories or Rathlin Island (arrow Figure 1) on return ships to Tröndelag for a further migration into its former province of Jämtland [41]. Whether the two HD clusters in Jämtland and Västernorrland (Figure 2) have a similar origin is unknown.
HD haplotype studies showed however strong similarities between the two (19/20 families had the haplotype 7A in common), although the haplotype was considered incor-rect according to recent studies [42–44]. This would mean a common ancestor beyond the 15th and the 16th century (Figure 6).
A Viking origin was previously discussed when the former Norwegian province of Bohuslän on the Swedish west coast was shown to carry Wilson´s disease in a HFE p.C282Y family [45,46].
6.3. Will Huntington´s disease be with us for many years?
According to a recent registry study of inpatients with HD (G10.9) in Jämtland, this region (Figure 1) has a four times higher prevalence than the central region (Uppsala). The annual average of inpatients with HD in Jämtland was 1.5/100000 [15].
By using the same statistical database available at http://www.socialstyrelsen.se/ statistics/statisticaldatabase, we found a similarly high frequency of 1.76/100000 in the region of Västernorrland (including the Mo river population). These frequencies are the highest in the country and are in agreement with the cluster of HD in Sweden, reminding us that “Huntington’s disease will be with us for many years” [46].
Today, the church records are digitally available, handwritten documents were easy to read, but the column showing causes of death often were found empty before 1918 (except suicide). “Danssjuka” (Swedish word for Huntington´s disease) was rarely reported (n = 3). “Skaksjuka” or shaking disease was reported once, “nervosity” or “nervous disease” in agreement with as reported by Almqvist et al.
We believe that HD was well known by the priests and being registered as “insanity or fool” in the parish catechetical 1908–1923 registry of “sinnessjuka och idioter”. The Figure 4 pedigree included 29 such affected (56.9 Percent) individuals and the Figure 6 pedigree shows all 51 in the registry.
This is not a proof for all having HD but gives support for Huntington´s disease being considerably more common than described, in agreement with other studies. While Wilson patients of today can be saved by an effective treatment such a fortune cannot yet be given to HD sufferers despite hard ongoing efforts.
Recently, several HD therapies are at trial stages, with a target-specific drug develop-ment focussed on the mechanisms related to mutant Huntingtin (HTT) protein.
Examples include Tominersen and Wave Life Sciences’ WVE-120101/WVE-120102 are experimental antisense oligonucleotides (ASOs) designed to treat Huntington’s dis-ease (HD) by reducing toxic huntingtin protein. Both faced significant setbacks, with PRECISION-HD trials for WVE-120101/WVE-120102 discontinued in 2021 due to lack of significant mutant huntingtin reduction. Splicing modifiers and microRNA molecules that aim to reduce the levels of mutant HTT protein, the therapeutic landscape continues to expand with various trials currently under development to document proof-of-concept and safety/tolerability. We totally agree with Peter S Harper who described Huntington´s disease as one of the most important genetic disorders of adult life [40] and also with Nancy Wexler who spent most of her life to support Huntington patients [47]. There are strong ongoing efforts to find a cure for HD[12]. Hopefully a treatment is within reach in the near future.
7. Ethical questions
Ethical questions may be raised because Huntington´s disease is still a feared disor-der, however none of the HD members are still alive to be asked for a permission. We understand that HD families of today may be more interested in the future of their children than their history. However, matters will be changed hopefully in the near future, when a therapy for HD is available.
8. Acknowledgment
The study was supported by a grant from Göteborg´s Medical Society, who had no interest in the study design or its writing. RR-C would like to pay a tribute to Professor Peter S Harper for his mentorship and who showed her the way to enter a “World of Genetics”. We are grateful to Dr Matts Andersson, the WD patient’s doctor who let us know about her and also about her parent’s interest in the family history, Professors Martin Stenson and Herman Nilsson-Ehle of Sahlgrenska University Hospital for their encouragement to publish Dr Olsson’s final contribution. We are also grateful to Dr Lars Olof Andersson who informed us about the new HD family and to Mrs Marie Fahlén and Mr Ronnie Sundin at regionarkivet@rvn.se for help with patient´s records of the past and Dr Subhojit Chakraborty editing the manuscript.
9. Authors contributions
KSO and RR-C wrote the manuscript and performed the pedigree analyses using church records obtained by the genealogist OS. Unfortunately, KSO passed away in August 2025. OW participated in the WD studies, while RR-C performed the DNA analyses and identified haplotypes for the HFE and HD studies. OW, SRC, the manuscript. All authors, except KSO and OS, read and approved the final manuscript. RR-C made only very limited changes to the final version of the manuscript.
References:
- Einarsdottir, E.; Egerbladh, I.; Beckman, L.; Holmberg, D.; Escher, S.A. The genetic population structure of northern Sweden and its implications for mapping genetic diseases. Hereditas 2007, 144, 171–180.
- Bittles, A.H.; Egerbladh, I. The influence of past endogamy and consanguinity on genetic disorders in northern Sweden. Ann. Hum. Genet. 2005, 69, 549–558.
- Ritter, B.; Säfwenberg, J.; Olsson, K.S. HLA as a marker of the hemochromatosis gene in Sweden. Hum. Genet. 1984, 68, 62–66.
- Olsson, K.S.; Ritter, B.; Hansson, N.; Raha-Chowdhury, R. HLA haplotype map of river valley populations with hemochromatosis traced through five centuries in Central Sweden. Eur. J. Haematol. 2008, 81, 36–46.
- Olsson, K.S.; Wålinder, O.; Kindmark, A.; Williams, R. Common local founder effects for Wilson's disease and hereditary hemochromatosis; mutation studies of a large family. Scand. J. Gastroenterol. 2012, 47, 1014–1020.
- Winbo, A.; Diamant, U.B.; Rydberg, A.; Persson, J.; Jensen, S.M.; Stattin, E.L. Origin of the Swedish long QT syndrome Y111C/KCNQ1 founder mutation. Heart Rhythm 2011, 8, 541–547.
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am. Heart J. 1957, 54, 59–68.
- Olsson, K.S.; Wålinder, O.; Jansson, U.; Wilbe, M.; Bondeson, M.L.; Stattin, E.L.; Raha-Chowdhury, R.; Williams, R. Common founder effects of hereditary hemochromatosis, Wilson's disease, the long QT syndrome and autosomal recessive deafness caused by two novel mutations in the WHRN and TMC1 genes. Hereditas 2017, 154, 16.
- Sjögren, T. Vererbungsmedizinische Untersuchungen über Huntingtons Chorea in einer schwedischen Bauernpopulation. Z. Menschl. Vererb. Konstitutionsl. 1936, 19, 131–165.
- Holmgren, G.; Almqvist, E.W.; Anvret, M.; Conneally, P.M.; Hobbs, W.; Mattsson, B.; Wahlström, J.; Winblad, B.; Gusella, J.F. Linkage of G8 (D4S10) in two Swedish families with Huntington's disease. Clin. Genet. 1987, 32, 289–294.
- Almqvist, E.; Andrew, S.; Theilmann, J.; Goldberg, Y.P.; Zeisler, J.; Drugge, U.; Grandell, U.; Tapper-Persson, M.; Winblad, B.; Hayden, M.R. Geographical distribution of haplotypes in Swedish families with Huntington's disease. Hum. Genet. 1994, 94, 124–128.
- Estevez-Fraga, C.; Flower, M.D.; Tabrizi, S.J. Therapeutic strategies for Huntington's disease. Curr. Opin. Neurol. 2020, 33, 508–518.
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983, 306, 234–238.
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993, 72, 971–983.
- Roos, A.K.; Wiklund, L.; Laurell, K. Discrepancy in prevalence of Huntington's disease in two Swedish regions. Acta Neurol. Scand. 2017, 136, 511–515.
- Sjöberg, B. Rötter i Anundsjö--en bygd Nolaskogs; Bengt Sjöberg Förlag: Lidingö, Sweden, 1999.
- Wilson, S.A.K. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912, 34, 295–509.
- Zanko, A.; Abrams, L. Case report: concurrent Wilson disease and Huntington disease: lightning can strike twice. J. Genet. Couns. 2015, 24, 40–45.
- Wexler, N.S.; U.S.-Venezuela Collaborative Research Project. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503.
- Paradisi, I.; De Freitas, L.; Arias, S. Most frequent mutation c.3402delC (p.Ala1135GlnfsX13) among Wilson disease patients in Venezuela has a wide distribution and two old origins. Eur. J. Med. Genet. 2015, 58, 59–65.
- Haines, J.L.; Ozelius, L.; St George-Hyslop, P.; Wexler, N.S.; Gusella, J.F.; Conneally, P.M. Partial linkage map of chromosome 13q in the region of the Wilson disease and retinoblastoma genes. Genet. Epidemiol. 1988, 5, 375–380.
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350.
- Paradisi, I.; Hernández, A.; Arias, S. Huntington disease mutation in Venezuela: Age of onset, haplotype analyses and geographic aggregation. J. Hum. Genet. 2008, 53, 127–135.
- Wexler, N.S.; Young, A.B.; Tanzi, R.E.; et al. Homozygotes for Huntington's disease. Nature 1987, 326, 194–197.
- Coffey, A.J.; Durkie, M.; Hague, S.; et al. A genetic study of Wilson's disease in the United Kingdom. Brain 2013, 136, 1476–1487.
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.; Smeeth, L. The prevalence of Huntington's disease. Neuroepidemiology 2016, 46, 144–153.
- Weydt, P.; Dupuis, L.; Petersen, Å. Thermoregulatory disorders in Huntington disease. Handb. Clin. Neurol. 2018, 157, 761–775.
- Austerlitz, F.; Heyer, E. Social transmission of reproductive behavior increases frequency of inherited disorders in a young-expanding population. Proc. Natl. Acad. Sci. USA 1998, 95, 15140–15144.
- Blum, M.G.B.; Heyer, E.; François, O.; Austerlitz, F. Matrilineal fertility inheritance detected in hunter-gatherer populations using the imbalance of gene genealogies. PLoS Genet. 2006, 2, e122.
- Harper, P.S. Genetic variation in Wales. J. R. Coll. Physicians Lond. 1976, 10, 321–332.
- Walker, D.A.; Harper, P.S.; Newcombe, R.G.; Davies, K. Huntington's chorea in South Wales: Mutation, fertility, and genetic fitness. J. Med. Genet. 1983, 20, 12–17.
- Morrison, P.J.; Johnston, W.P.; Nevin, N.C. The epidemiology of Huntington's disease in Northern Ireland. J. Med. Genet. 1995, 32, 524–530.
- Huntington, G. On chorea. Med. Surg. Rep. 1872, 26, 320–321.
- Bolt, J.M.W. Huntington's chorea in the West of Scotland. Br. J. Psychiatry 1970, 116, 259–270.
- Scriver, C.R. Human genetics: Lessons from Quebec populations. Annu. Rev. Genomics Hum. Genet. 2001, 2, 69–101.
- Chong, J.X.; Ouwenga, R.; Anderson, R.L.; Waggoner, D.J.; Ober, C. A population-based study of autosomal-recessive disease-causing mutations in a founder population. Am. J. Hum. Genet. 2012, 91, 608–620.
- Seielstad, M.T.; Minch, E.; Cavalli-Sforza, L.L. Genetic evidence for a higher female migration rate in humans. Nat. Genet. 1998, 20, 278–280.
- Ikonen, E.; Ignatius, J.; Norio, R.; Palo, J.; Peltonen, L. Huntington disease in Finland: A molecular and genealogical study. Hum. Genet. 1992, 89, 275–280.
- Cassidy, L.M.; Martiniano, R.; Murphy, E.M.; et al. Neolithic and Bronze Age migration to Ireland and establishment of the insular Atlantic genome. Proc. Natl. Acad. Sci. USA 2016, 113, 368–373.
- Harper, P.S. The epidemiology of Huntington's disease. Hum. Genet. 1992, 89, 365–376.
- Margaryan, A.; Lawson, D.J.; Sikora, M.; Willerslev, E.; et al. Population genomics of the Viking world. Nature 2020.
- Gudmundsson, K.R. Prevalence and occurrence of some rare neurological diseases in Iceland. Acta Neurol. Scand. 1969, 45, 114–118.
- Bickford, J.A.; Ellison, R.M. The high incidence of Huntington's chorea in the Duchy of Cornwall. J. Ment. Sci. 1953, 99, 291–294.
- Lyon, R.L.L. Huntington's chorea in the Moray Firth area. Br. Med. J. 1962, 1, 1301–1306.
- Olsson, K.S.; Konar, J.; Dufva, I.H.; Ricksten, A.; Raha-Chowdhury, R. Was the C282Y mutation an Irish Gaelic mutation that the Vikings helped disseminate? HLA haplotype observations of hemochromatosis from the west coast of Sweden. Eur. J. Haematol. 2011, 86, 75–82.
- Rodríguez-Varela, R.; Götherström, A.; et al. The genetic history of Scandinavia from the Roman Iron Age to the present. Cell 2023, 186, 32–46.
- Fischbeck, K.H.; Wexler, N.S. Oligonucleotide treatment for Huntington's disease. N. Engl. J. Med. 2019, 380, 2373–2374.